


La línea Life Science está especializada en el desarrollo y la comercialización de servicios y productos biotecnológicos para la investigación en Ciencias de la Vida.
Nuestra tecnología, equipo humano, instalaciones y socios corporativos focalizan todo su esfuerzo en ayudarle a avanzar en sus proyectos de investigación.

Ingeniería genética
Sintetizamos sus genes de novo, y sus oligos, ajustándonos por completo a sus necesidades y guiándole siempre a través de nuestros expertos. Encontrará un amplio rango de posibilidades dentro de este servicio.
Adicionalmente, contamos con una relación calidad-precio muy competitiva en nuestros servicios de secuenciación de última tecnología, tanto Sanger como NGS.
Síntesis de péptidos personalizados
Nuestro servicio de síntesis de péptidos está diseñado para ofrecerle precios altamente competitivos sin comprometer la calidad del servicio ni los tiempos de entrega. Nos adaptamos a sus necesidades específicas, sin límite de tamaño en la secuencia de aminoácidos, garantizando cantidad, pureza y todo tipo de modificaciones.

Producción de proteínas recombinantes
Nuestro equipo de expertos cuenta con más de diez años de experiencia en expresión de proteínas recombinantes. Nuestro laboratorio trabaja con la tecnología más avanzada para ofrecer cinco sistemas de expresión con diferentes rendimientos y posibilidades de modificaciones postraduccionales: E. coli, B. subtilis, Levadura, Baculovirus, Células de mamífero.

Producción de anticuerpos
Nuestro propósito es ofrecer un servicio integral, de producción anticuerpos policlonales y monoclonales totalmente personalizado para el cliente. Le guiamos desde el diseño del antígeno hasta el anticuerpo purificado.
Ofrecemos una gran diversidad de especies productoras, servicio de criopreservación y expansión del hibridoma y test de micoplasma, entre otros.
Ingeniería genética
Sintetizamos sus genes de novo, y sus oligos, ajustándonos por completo a sus necesidades y guiándole siempre a través de nuestros expertos. Encontrará un amplio rango de posibilidades dentro de este servicio.
Adicionalmente, contamos con una relación calidad-precio muy competitiva en nuestros servicios de secuenciación de última tecnología, tanto Sanger como NGS.
Síntesis de péptidos personalizados
Nuestro servicio de síntesis de péptidos está diseñado para ofrecerle precios altamente competitivos sin comprometer la calidad del servicio ni los tiempos de entrega. Nos adaptamos a sus necesidades específicas, sin límite de tamaño en la secuencia de aminoácidos, garantizando cantidad, pureza y todo tipo de modificaciones.
Producción de proteínas recombinantes
Nuestro equipo de expertos cuenta con más de diez años de experiencia en expresión de proteínas recombinantes. Nuestro laboratorio trabaja con la tecnología más avanzada para ofrecer cinco sistemas de expresión con diferentes rendimientos y posibilidades de modificaciones postraduccionales: E. coli, B. subtilis, Levadura, Baculovirus, Células de mamífero.
Producción de anticuerpos
Nuestro propósito es ofrecer un servicio integral, de producción anticuerpos policlonales y monoclonales totalmente personalizado para el cliente. Le guiamos desde el diseño del antígeno hasta el anticuerpo purificado.
Ofrecemos una gran diversidad de especies productoras, servicio de criopreservación y expansión del hibridoma y test de micoplasma, entre otros.
Ofrecemos soluciones integradas
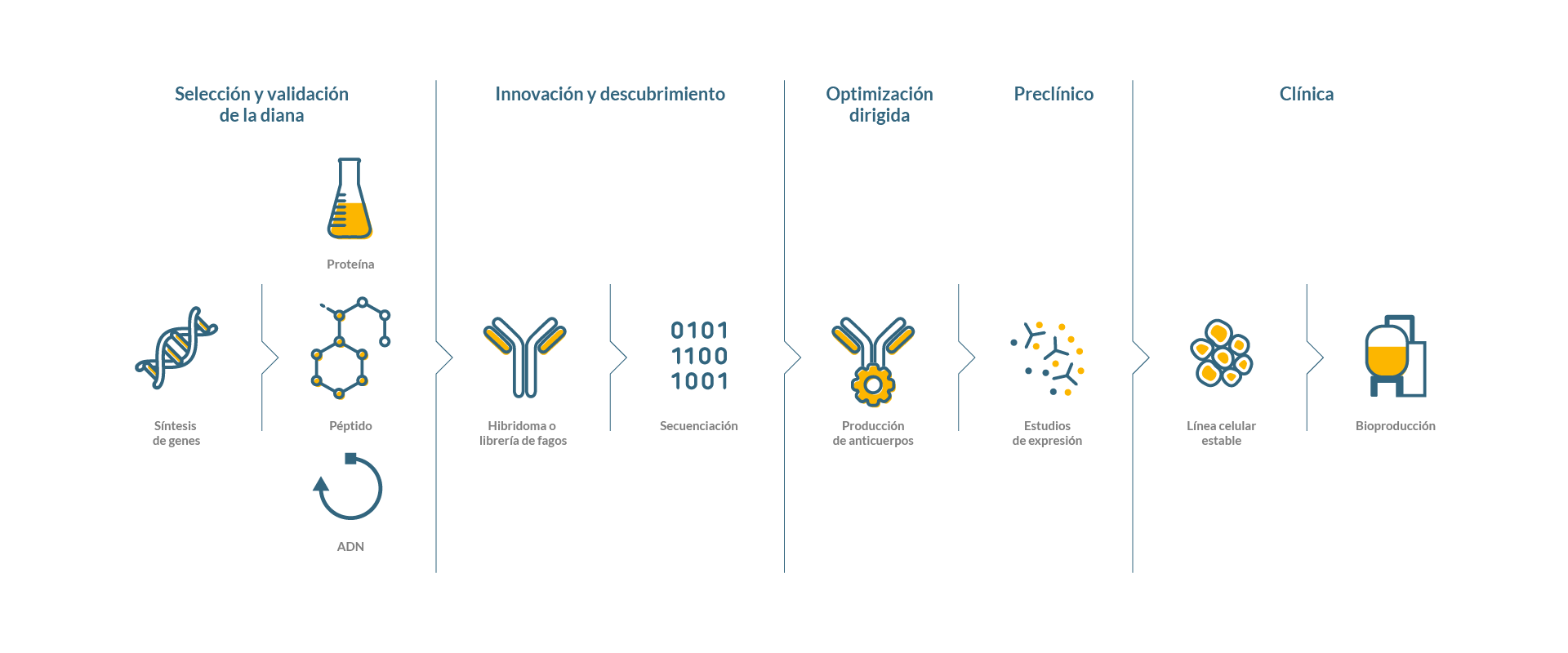
Ofrecemos soluciones integradas
